RBFE calculations of a Protein-Membrane System#
This tutorial walks you through the process of setting up a relative binding free energy (RBFE) simulation campaign for a membrane protein system using OpenFE.
RBFE calculations for membrane proteins require additional preparation compared to soluble protein systems. For soluble proteins, solvation and box setup are handled automatically within the OpenFE protocol. In contrast, membrane protein systems must be provided as fully built, solvated, and pre-equilibrated systems, including correctly defined box vectors. You can prepare these systems using tools such as packmol-memgen, CHARMM-GUI, or Maestro.
Note: This tutorial focuses on importing inputs prepared with Maestro. However, the overall procedure is applicable to systems generated with other preparation tools.
Warning: The membrane must be aligned with the coordinate axes to ensure correct pressure coupling with a membrane barostat during the simulation.
[1]:
import numpy as np
import openmm
import openfe
from openfe import ProteinMembraneComponent, ProteinComponent
from openff.units import unit as offunit
from rdkit import Chem
import logging
logger = logging.getLogger()
logger.setLevel(logging.ERROR)
(Optional) Combining System Components into a Single PDB File#
Maestro exports solvated protein-membrane systems in a non-standard PDB format that OpenMM cannot directly read. To simplify the workflow, it is often easier to save different parts of the system separately (e.g. the protein, lipids, and water) and then combine them into a single PDB file.
Using OpenMM’s Modeller, the components can be merged as follows:
[2]:
# Specifying input and output filenames
pdb_lipids = 'a2a/a2a_lipids.pdb'
pdb_protein = 'a2a/a2a_protein.pdb'
pdb_water = 'a2a/a2a_water.pdb'
pdb_complex = 'a2a/a2a_complex.pdb' # specify the output filename
Step 1: Load each component#
We start by loading the protein, lipids, and water as ProteinComponent objects from their respective PDB files:
[3]:
protein = ProteinComponent.from_pdb_file(pdb_protein)
lipids = ProteinComponent.from_pdb_file(pdb_lipids)
water = ProteinComponent.from_pdb_file(pdb_water)
/Users/atravitz/micromamba/envs/openfe_env/lib/python3.13/site-packages/gufe/vendor/pdb_file/pdbstructure.py:465: UserWarning: WARNING: two consecutive residues with same number (ATOM 3205 N GLU A 219 9.932 6.197 -38.597 1.00 0.00 N , ATOM 3220 HH33 ACE A 219 11.901 4.558 -37.817 0.00 0.00 H )
warnings.warn(
Step 2: Merge components using OpenMM’s Modeller and save the merged system to a single PDB file#
We create a Modeller with the protein first, then add lipids and water. This ensures all atoms are included in a single system. Finally, we write the combined system to a new PDB file that is ready for simulation.
[4]:
# Create modeller using the protein inputs
modeller = openmm.app.Modeller(protein.to_openmm_topology(), protein.to_openmm_positions())
# Add the lipids
modeller.add(lipids.to_openmm_topology(), lipids.to_openmm_positions())
# Add the solvent
modeller.add(water.to_openmm_topology(), water.to_openmm_positions())
# Write out the new topology and positions
mergedTopology = modeller.topology
mergedPositions = modeller.positions
with open(pdb_complex, "w") as f:
openmm.app.pdbfile.PDBFile.writeFile(modeller.topology, modeller.positions, f)
Using the openfe CLI with membrane systems#
For basic membrane support, you may use the CLI command openfe plan-rbfe-network with the --protein-membrane argument in place of the usual --protein argument. You will still need to have a fully built, solvated, and pre-equilibrated system that includes box vectors in a PDB or PDBx/mmCIF file when using the CLI.
For the full functionality available in the openfe Python API, continue this tutorial.
Loading the system as a ProteinMembraneComponent with Periodic Box Vectors#
The ``ProteinMembraneComponent`` requires periodic box vectors to define the simulation box. There are several ways to provide these vectors:
- ``CRYST`` record in the PDB fileIf your PDB file includes a CRYST record (common in files exported from Maestro or other modeling tools), OpenMM can automatically read the box vectors from it.
- Manually specifying box vectorsYou can provide the box vectors explicitly in OpenMM format (see below).
- Inferring from atomic positionsBox vectors can be estimated from the atomic coordinates in the PDB file. Caveat: This approach can be inaccurate if the PDB comes from a previous simulation and some atoms are positioned in periodic images.
Maestro-specific note: When merging multiple components exported from Maestro, CRYST records are not currently preserved in the combined output PDB file. If one of the original input files contains valid CRYST information (for example, a2a_water.pdb in this case), you can load that file directly as a ProteinMembraneComponent and extract the box vectors from it, as shown below.
Option 1: CRYST record in the PDB file#
Box vectors are automatically recognized when stored in the CRYST record. In our case, only the a2a_water.pdb file had a CRYST record.
[5]:
a2a_cryst = "a2a/a2a_water.pdb"
[6]:
system_with_box = openfe.ProteinMembraneComponent.from_pdb_file(a2a_cryst)
[7]:
box_vector = system_with_box.box_vectors
box_vector
[7]:
| Magnitude | [[6.9587 0.0 0.0] [0.0 5.9164 0.0] [0.0 0.0 9.2692]] |
|---|---|
| Units | nanometer |
Option 2: Manually provide box vectors#
Some membrane setup tools (e.g. CHARMM-GUI or Maestro) store the periodic box vectors in the outputs of the membrane-building step or a prior equilibration simulation. In these cases, you can manually supply the box vectors to OpenFE.
Box vectors must be provided as a NumPy array with OpenFF units. The vectors must be specified in their reduced form, as required by OpenMM.
For details on reduced box vectors and periodic boundary conditions, see the OpenMM documentation: https://docs.openmm.org/latest/userguide/theory/05_other_features.html#periodic-boundary-conditions
[8]:
box_vector = np.array([
[6.9587, 0.0, 0.0],
[0.0, 5.9164, 0.0],
[0.0, 0.0, 9.2692]
]) * offunit.nanometer
[9]:
membrane_protein_user_box = openfe.ProteinMembraneComponent.from_pdb_file(pdb_complex, box_vectors=box_vector)
[10]:
membrane_protein_user_box.box_vectors
[10]:
| Magnitude | [[6.9587 0.0 0.0] [0.0 5.9164 0.0] [0.0 0.0 9.2692]] |
|---|---|
| Units | nanometer |
Option 3: Infer box vectors from atomic positions#
As a fallback, box vectors can be estimated from the atomic coordinates in the PDB file. An orthorhombic box is constructed by taking the minimum and maximum coordinates along each axis and expanding the resulting bounding box by an optional padding.
Caveat: When merging multiple components exported from Maestro, CRYST records are not currently preserved in the combined output PDB file. If one of the original input files contains valid CRYST information (for example, a2a_water.pdb in this case), you can load that file directly as a ProteinMembraneComponent and extract the box vectors from it, as shown below.
This approach is convenient when no box information is available, but should generally be used as a last resort.
[11]:
membrane_protein_infer_box = openfe.ProteinMembraneComponent.from_pdb_file(pdb_complex, infer_box_vectors=True)
/Users/atravitz/micromamba/envs/openfe_env/lib/python3.13/site-packages/gufe/components/solvatedpdbcomponent.py:230: UserWarning: Box vectors were inferred from the atomic coordinates.
Note: This heuristic assumes that the coordinates reflect the true periodic unit cell. It may produce incorrect box dimensions for structures that were post-processed (e.g., unwrapped after MD).
Inferred box vectors: [[9.445200000000002 0.0 0.0] [0.0 8.196100000000001 0.0] [0.0 0.0 9.8092]] nanometer
Applied padding: 0.2 nanometer
warnings.warn(
This box is much bigger than the other boxes since PBC is not explicitly handled in infer_box_vector.
[12]:
membrane_protein_infer_box.box_vectors
[12]:
| Magnitude | [[9.445200000000002 0.0 0.0] [0.0 8.196100000000001 0.0] [0.0 0.0 9.8092]] |
|---|---|
| Units | nanometer |
[13]:
# The density is quite low for this case since the box is too big
membrane_protein_infer_box.density
[13]:
Validating Your ProteinMembraneComponent#
After adding periodic box vectors, it’s a good practice to validate your ProteinMembraneComponent. This helps catch common issues early, specifically:
- Number of waters:Ensures that the system contains at least the specified
min_waters. If there are too few, it may indicate missing solvent.Default: 50 - System density:Checks that the density is above
min_density. A very low density often signals incorrect box vectors or missing solvent.Default: 700 g/L
If any of these checks fail, the validate method will raise a ValueError, indicating that the system may be missing solvent or have incorrect box vectors.
[14]:
# Our created `ProteinMembraneComponent` should not give any errors
membrane_protein_user_box.validate()
[15]:
# The `ProteinMembraneComponent` with inferred box vectors should give an error
from gufe.components.errors import ComponentValidationError
try:
membrane_protein_infer_box.validate()
except ComponentValidationError as e:
print(e)
ProteinMembraneComponent validation failed:
- Estimated system density is very low.
Density: 519.107 gram / liter (expected ≥ 0.7 gram / milliliter). This usually indicates missing solvent or incorrect box vectors.
This usually indicates missing solvent or incorrect box vectors.
[16]:
# The pure protein, without lipids and waters, should give errors
only_protein = openfe.ProteinMembraneComponent.from_pdb_file(pdb_protein, box_vectors=box_vector)
try:
only_protein.validate()
except ComponentValidationError as e:
print(e)
ProteinMembraneComponent validation failed:
- Estimated system density is very low.
Density: 141.802 gram / liter (expected ≥ 0.7 gram / milliliter). This usually indicates missing solvent or incorrect box vectors.
- Only 0 water molecules detected (expected ≥ 50).
This usually indicates missing solvent or incorrect box vectors.
Loading the ligands and creating a LigandNetwork#
Next, we load the small molecules for which we want to calculate free energies. In this example, the ligands are stored in an SDF file containing multiple molecules. We can load them using RDKit’s SDMolSupplier and pass them to OpenFE:
[17]:
ligands_sdf = Chem.SDMolSupplier("a2a/ligands_am1bcc.sdf", removeHs=False)
# Now pass these to form a list of Molecules
ligands = [openfe.SmallMoleculeComponent(sdf) for sdf in ligands_sdf]
The LigandNetwork is then created using three main components:
Atom Mapper – Proposes potential atom mappings between ligand pairs.
Scorer – Evaluates the quality of each proposed mapping.
Network Planner – Generates the network itself.
[18]:
mapper = openfe.setup.KartografAtomMapper()
scorer = openfe.lomap_scorers.default_lomap_score
network_planner = openfe.ligand_network_planning.generate_lomap_network
[19]:
ligand_network = network_planner(
ligands=ligands,
mappers=[mapper],
scorer=scorer
)
[20]:
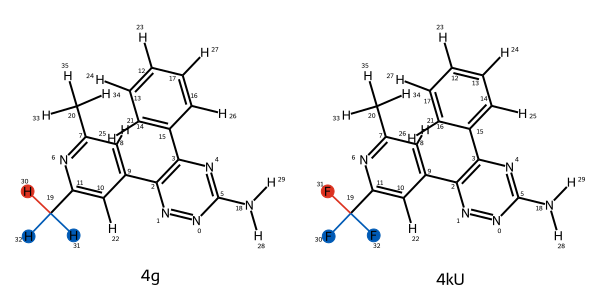
# get the first edge; it automatically displays in a Jupyter notebook
mapping = next(iter(ligand_network.edges))
[21]:
mapping
Creating the AlchemicalNetwork#
The ``AlchemicalNetwork`` contains all the information needed to run a complete RBFE campaign. Each Transformation represents one alchemical mutation between a pair of ligands in a specific environment (either solvent or protein-membrane complex).
In this example, we loop over all ligand mappings, and for each mapping, we loop over the legs (solvent and complex) to create the corresponding Transformation objects.
The default settings apply the AMBER lipid17 forcefield to the lipids. Additional lipid forcefields from OpenMMForcefields, such as AMBER lipid21, can also be added (note: not yet tested with this Protocol).
In this example, we use ``_adaptive_settings`` in RelativeHybridTopologyProtocol to generate protocol settings based on the system composition:
Complex leg (protein-membrane)
Uses a
MonteCarloMembraneBarostatfor appropriate membrane pressure coupling.
Solvent leg
Uses a standard
MonteCarloBarostatfor bulk solvent pressure regulation.
[22]:
from openfe.protocols.openmm_rfe import RelativeHybridTopologyProtocol
transformations = []
for mapping in ligand_network.edges:
for leg in ['solvent', 'complex']:
if leg == 'solvent':
sysA_dict = {'ligand': mapping.componentA,
'solvent': openfe.SolventComponent()}
sysB_dict = {'ligand': mapping.componentB,
'solvent': openfe.SolventComponent()}
if leg == 'complex':
sysA_dict = {'ligand': mapping.componentA,
'protein': membrane_protein_user_box}
sysB_dict = {'ligand': mapping.componentB,
'protein': membrane_protein_user_box}
# we don't have to name objects, but it can make things (like filenames) more convenient
system_a = openfe.ChemicalSystem(sysA_dict, name=f"{mapping.componentA.name}_{leg}")
system_b = openfe.ChemicalSystem(sysB_dict, name=f"{mapping.componentB.name}_{leg}")
prefix = "rbfe_"
protocol_settings = RelativeHybridTopologyProtocol.default_settings()
protocol_settings.protocol_repeats = 1
protocol = RelativeHybridTopologyProtocol(
settings=RelativeHybridTopologyProtocol._adaptive_settings(
stateA=system_a,
stateB=system_b,
mapping=mapping,
initial_settings=protocol_settings,
),
)
transformation = openfe.Transformation(
stateA=system_a,
stateB=system_b,
mapping=mapping,
protocol=protocol, # use protocol created above
name=f"{prefix}{system_a.name}_{system_b.name}"
)
transformations.append(transformation)
network = openfe.AlchemicalNetwork(transformations)
[23]:
# A solvent leg transformation uses the `MonteCarloBarostat`
transformations[-2].protocol.settings.integrator_settings
{'barostat': 'MonteCarloBarostat',
'barostat_frequency': {'unit': 'timestep', 'val': 25.0},
'constraint_tolerance': 1e-06,
'langevin_collision_rate': {'unit': '1 / picosecond', 'val': 1.0},
'n_restart_attempts': 20,
'reassign_velocities': False,
'remove_com': False,
'surface_tension': {'unit': 'bar * nanometer', 'val': 0},
'timestep': {'unit': 'femtosecond', 'val': 4.0}}
[24]:
# A complex leg transformation uses the `MonteCarloMembraneBarostat`
transformations[-1].protocol.settings.integrator_settings
{'barostat': 'MonteCarloMembraneBarostat',
'barostat_frequency': {'unit': 'timestep', 'val': 25.0},
'constraint_tolerance': 1e-06,
'langevin_collision_rate': {'unit': '1 / picosecond', 'val': 1.0},
'n_restart_attempts': 20,
'reassign_velocities': False,
'remove_com': False,
'surface_tension': {'unit': 'bar * nanometer', 'val': 0},
'timestep': {'unit': 'femtosecond', 'val': 4.0}}
Writing the Transformations to disk#
We’ll write out each transformation to disk, so that they can be run independently using the openfe quickrun command:
[25]:
import pathlib
# first we create the directory
transformation_dir = pathlib.Path("transformations")
transformation_dir.mkdir(exist_ok=True)
# then we write out each transformation
for transformation in network.edges:
transformation.to_json(transformation_dir / f"{transformation.name}.json")
Each of these individual .json files contains a Transformation, which contains all the information to run the calculation. These could be farmed out as individual jobs on a HPC cluster. For details on running them, please follow from the section on running simulations in the CLI tutorial (https://docs.openfree.energy/en/latest/tutorials/rbfe_cli_tutorial.html)