Planning a Ligand Network#
For an introduction to LigandNetworks, see User Guide: Defining the Ligand Network.
Load the Ligands#
This cookbook assumes you have already loaded a collection of SmallMoleculeComponent objects into an iterable called ligands.
For more information, see the Loading Small Molecules Cookbook.
[6]:
%matplotlib inline
from rdkit import Chem
import openfe
supplier = Chem.SDMolSupplier("assets/somebenzenes.sdf", removeHs=False)
ligands = [openfe.SmallMoleculeComponent(mol) for mol in supplier]
ligands
[6]:
[SmallMoleculeComponent(name=benzene),
SmallMoleculeComponent(name=toluene),
SmallMoleculeComponent(name=phenol),
SmallMoleculeComponent(name=benzonitrile),
SmallMoleculeComponent(name=anisole),
SmallMoleculeComponent(name=benzaldehyde),
SmallMoleculeComponent(name=styrene)]
Choose an atom mapper#
The AtomMapper will generate the AtomMappings that relate two small molecules and serve as the edges of our LigandNetwork. Atom mapper classes inherit from LigandAtomMapper.
Each atom mapper suggests one or more mappings, which can be scored seperately. As a result, multiple atom mappers can be used to generate additional mappings for network planning.
[2]:
from openmm import unit
lomap_mapper = openfe.setup.LomapAtomMapper(
time=20, # Time out if MCS algorithm takes 20 seconds
threed=True, # Use atom positions to prune symmetric mappings
max3d=1.0, # Forbid mapping between atoms more than 1.0 Å apart
element_change=True, # Allow mappings that change an atoms element
seed='', # Empty SMARTS string causes MCS search to start from scratch
shift=False, # Keep pre-aligned atom positions for 3D position checks
)
kartograf_mapper = openfe.setup.KartografAtomMapper()
# to demonstrate passing multiple atom mappers to the planner
mappers = [lomap_mapper, kartograf_mapper]
Choose a scoring function#
Many ligand network planners require a Mapping Scorer to assign scores (i.e. edge weights) to each of the transformations (i.e. network edges) to be used as an estimation of the quality of the mapping.
A Mapping Scorer is a function that takes an atom mapping and returns a score between 0.0, very hard to compute, and 1.0 for easy to compute.
In other words, transformations that are expected to converge quickly without introducing artifacts are scored highly, while more drastic or risky transformations are scored lower.
Keep in mind that edge scorers are fallible and a score is only the anticipated difficulty of a transformation to be used for network creation.
[3]:
# use LOMAP scorer with defaults
scorer = openfe.lomap_scorers.default_lomap_score
Plan the network#
Finally, pass the ligands, mapper and scorer to a planner to generate the network. A planner takes these three arguments (and perhaps some more) and returns a LigandNetwork. Each planner implements a particular strategy for structuring a network. Different strategies may balance the precision-performance trade-off differently, provide edge redundancy for troubleshooting, or be more or less appropriate for a given set of ligands:
[7]:
# Choose the network that minimises the sum of scores
mst_network = openfe.ligand_network_planning.generate_minimal_spanning_network(
ligands=ligands,
mappers=mappers,
scorer=scorer,
);
# Choose the lomap network that adds some closed ligand cycles
lomap_network = openfe.ligand_network_planning.generate_lomap_network(
molecules=ligands,
mappers=mappers,
scorer=scorer,
);
# # Connect each ligand to a central ligand
radial_network = openfe.ligand_network_planning.generate_radial_network(
ligands=ligands[1:],
central_ligand=ligands[0],
mappers=mappers,
scorer=scorer,
);
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
/Users/atravitz/micromamba/envs/openfe-notebooks/lib/python3.13/site-packages/lomap/utils.py:20: DeprecationWarning: generate_lomap_network argument 'molecules' is deprecated, please use 'ligands' instead.
warnings.warn(deprecation_msg, DeprecationWarning)
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: Trying to remove edge 4-5 with similarity 0.670320
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 4-5
INFO: Trying to remove edge 3-4 with similarity 0.670320
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 3-4
INFO: Trying to remove edge 3-5 with similarity 0.704688
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 3-5
INFO: Trying to remove edge 3-6 with similarity 0.704688
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 3-6
INFO: Trying to remove edge 4-6 with similarity 0.704688
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 4-6
INFO: Trying to remove edge 2-3 with similarity 0.740818
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 2-3
INFO: Trying to remove edge 2-5 with similarity 0.740818
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 2-5
INFO: Trying to remove edge 2-6 with similarity 0.740818
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 2-6
INFO: Trying to remove edge 1-3 with similarity 0.778801
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 1-5 with similarity 0.778801
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 1-5
INFO: Trying to remove edge 1-6 with similarity 0.778801
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 1-6
INFO: Trying to remove edge 0-3 with similarity 0.818731
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 0-4 with similarity 0.818731
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 0-4
INFO: Trying to remove edge 0-5 with similarity 0.818731
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 0-6 with similarity 0.818731
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 1-4 with similarity 0.860708
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 0-1 with similarity 0.904837
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 0-1
INFO: Trying to remove edge 0-2 with similarity 0.904837
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 2-4 with similarity 0.904837
INFO: Rejecting edge deletion on cycle covering
INFO: Trying to remove edge 1-2 with similarity 0.951229
INFO: Checking edge deletion on distance-to-actives 0 vs 0
INFO: Removed edge 1-2
INFO: Trying to remove edge 5-6 with similarity 0.951229
INFO: Rejecting edge deletion on cycle covering
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
INFO: #################################
INFO: Map Heavy Atoms
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: #################################
INFO: Map Hydrogen Atoms:
INFO: #################################
INFO: Masking Atoms
INFO: Build Distance Matrix
INFO: Calculate Mapping
INFO: Find Maximal overlapping connected sets of mapped atoms
INFO: Filtering bond breaks
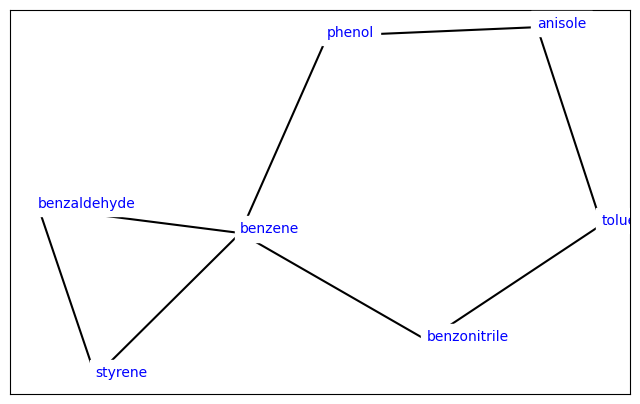
Visualize the Network#
For more ways to visualize a LigandNetwork, see Visualizing Ligand Networks.
[5]:
from openfe.utils.atommapping_network_plotting import plot_atommapping_network
plot_atommapping_network(lomap_network)
[5]: